[CAS NO. 136470-78-5] Abacavir (1592U89)
Please click "REQUEST A QUOTE" button if you need other sizes or custom synthesis
request a quote
If there is no stock, or you need other sizes or custom synthesis, please:
PRODUCTS SPECIFICATIONS [136470-78-5]
Store
Catalog
SLK-S5215
Brand
Selleck
CAS
136470-78-5
DESCRIPTION [136470-78-5]
Overview
| MDL | MFCD00903850 |
|---|---|
| Molecular Weight | 286.33 |
| Molecular Formula | C14H18N6O |
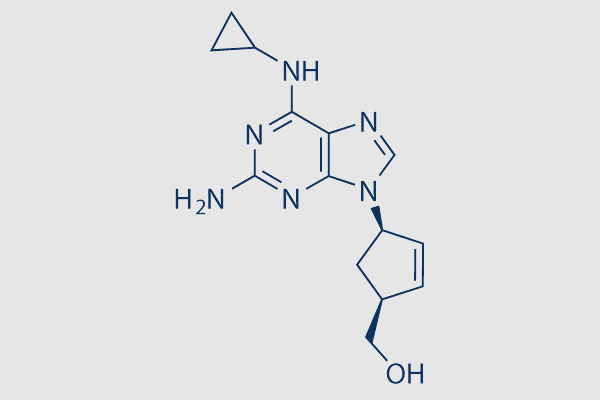
| SMILES | N(C1=C2C(N(C=N2)[C@@]3(C[C@H](CO)C=C3)[H])=NC(N)=N1)C4CC4 |
For research use only.
Storage
3 years,-20°C,powder
1 years,-80°C,in solvent
1 years,-80°C,in solvent
Shipping
Room temperature shipping(Stability testing shows this product can be shipped without any cooling measures.)
Preparing Stock Solutions
| 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.4925 mL | 17.4624 mL | 34.9247 mL |
| 5 mM | 0.6985 mL | 3.4925 mL | 6.9849 mL |
| 10 mM | 0.3492 mL | 1.7462 mL | 3.4925 mL |
| 50 mM | 0.0698 mL | 0.3492 mL | 0.6985 mL |
Description
Abacavir (1592U89, ABC) is a powerful used to treat HIV and AIDS.
Targets
| Reverse transcriptase [1] |
In vitro
Abacavir (ABC) exhibits potent in vitro antiviral activity against wild-type HIV-1 (IC50 4.0 μM, MT-4 cells). Abacavir induces chromosomal DSBs and thereby kills ATL cells but not non-HTLV-1-infected cells. Once abacavir is incorporated into the cells, it is phosphorylated in a unique stepwise anabolism to be converted to the triphosphated guanine analog carbovir (CBV) and then incorporated into host chromosomal DNA by replicative DNA polymerases, leading to premature termination of DNA replication, collapse of the replication fork, and DSB formation. Abacavir induces S/G2-phase arrest and apoptosis in ED-40515(−) cells, but not in Jurkat cells.
In vivo
Abacavir efficiently inhibits the growth of ATL cell xenografts in NOD/SCID mice. In adults, Abacavir is rapidly absorbed after oral administration, with peak concentrations occurring 0.63-1 hour after dosing. The absolute bioavailability of abacavir is approximately 83%. Abacavir pharmacokinetics are linear and doseproportional over the range of 300-1200 mg/day. The apparent volume of distribution of abacavir after intravenous administration is approximately 0.86 ± 0.15 L/kg, suggesting that abacavir is distributed to extravascular spaces. Binding to plasma proteins is about 50% and is independent of the plasma abacavir concentration. Abacavir is extensively metabolized by the liver; less than 2% is excreted as unchanged drug in the urine. Abacavir is primarily metabolized via two pathways, uridine diphosphate glucuronyltransferase and alcohol dehydrogenase, resulting in the inactive glucuronide metabolite and the inactive carboxylate metabolite. The terminal elimination half-life of abacavir is approximately 1.5 hours. The antiviral effect of abacavir is due to its intracellular anabolite, carbovirtriphosphate (CBV-TP). Abacavir is not significantly metabolized by cytochrome P450 (CYP) enzymes, nor does it inhibit these enzymes.
References
Synonyms
2-Cyclopentene-1-methanol, 4-[2-amino-6-(cyclopropylamino)-9H-purin-9-yl]-, (1S,4R)-
2-Cyclopentene-1-methanol, 4-[2-amino-6-(cyclopropylamino)-9H-purin-9-yl]-, (1S-cis)-
(1S,4R)-4-[2-Amino-6-(cyclopropylamino)-9H-purin-9-yl]-2-cyclopentene-1-methanol
Abacavir
1592U89
ABC
(-)-Abacavir
Powered by Arctom Copyright © 2026 Arctom. All rights
reserved.